1900 2039 (ext: 322; 339)
1900 2039 (ext: 322; 339) kd@ntt.edu.vn
kd@ntt.edu.vnNTTU – Phương pháp Molecular Docking sử dụng các tiến bộ trong tin học để sàng lọc ảo, mô tả và dự đoán các cấu trúc mới được cho là có hoạt tính sinh học cao. Những phương pháp sàng lọc rút ngắn thời gian và giảm chi phí trong quá trình nghiên cứu và phát triển thuốc. Hợp chất tự nhiên khi lựa chọn để nghiên cứu không bắt buộc có sẵn và việc thử nghiệm chúng chỉ là mô phỏng ảo, do đó không gây tốn kém về nguyên vật liệu. Từ đó với một chất bất kỳ cũng có thể được đánh giá thông qua sàng lọc ảo, tùy vào nghiên cứu, cơ sở dữ liệu hợp chất cho sàng lọc ảo có thể lên tới vài triệu hợp chất và toàn bộ có thể được phân tích chỉ qua một lần sàng lọc. Phương pháp sàng lọc thường được sử dụng các tương tác giữa thụ thể (Receptor) – hợp chất (Ligand) để tìm ra các hợp chất có cấu trúc được dự đoán liên kết với thụ thể tốt nhất thể hiện qua mức năng lượng ΔG thấp nhất [1].
Thông thường, mỗi loại thuốc mới được đưa ra thị trường phải tốn kém khoảng 800 triệu Euro và tốn thời gian 10–15 năm [2]. Trong khi đó, với các hệ thống máy tính nối mạng hiện đại (ví dụ tính toán lưới – Grid) thì hàng triệu cấu trúc có thể được sàng lọc ảo chỉ trong thời gian vài tuần [3].
Các sàng lọc in silico sử dụng các tương tác giữa Receptor – Ligand để tìm ra các hợp chất (Ligand) có cấu trúc được dự đoán liên kết với thụ thể tốt nhất – ở đây là có mức năng lượng ΔG thấp nhất (hình 1). Cấu trúc các protein đích ở mô hình 3 chiều (3D) đối với mỗi bệnh được cung cấp bởi các nhà sinh học, các ligand được phát triển dựa theo cấu trúc của các hợp chất hoá học, đặc biệt là các bộ khung cacbon đã được biết rõ ràng và có nguồn cung cấp, ngoài ra các sàng lọc này yêu cầu các phần mềm máy tính bản quyền và hệ thống máy tính với tốc độ rất nhanh [4].
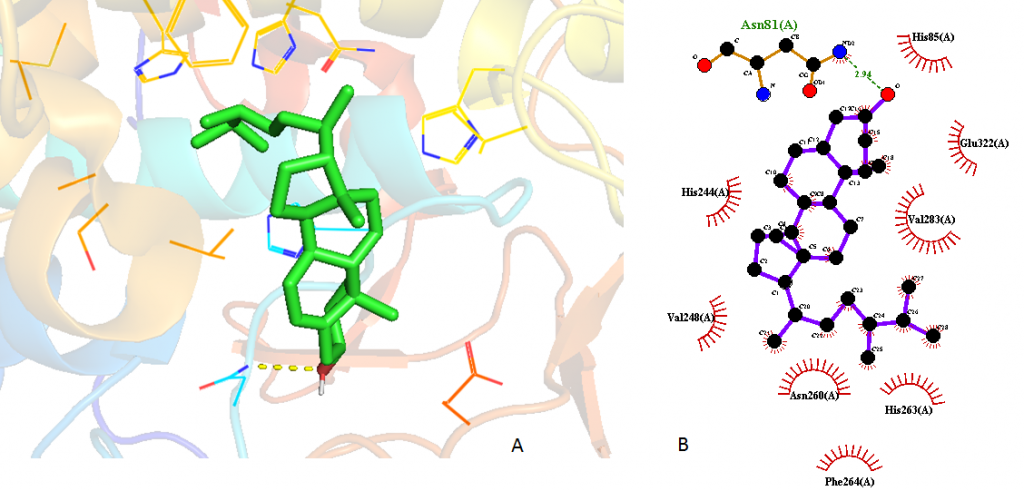
Hình ảnh 3D (trái) và 2D (phải) của Campesterol gắn kết với Tyrosinase
Phương pháp in silico docking được ứng dụng trong quá trình nghiên cứu và phát hiện thuốc. Phương pháp này được ứng dụng hầu hết các giai đoạn của quá trình nghiên cứu và phát hiện thuốc, từ việc tìm kiếm các hợp chất hóa học có tác dụng sinh học, đến tối ưu hóa cấu trúc các hợp chất này nhằm tăng hoạt tính sinh học, giảm độc tính, tăng các tính chất dược động học của thuốc, đến các giai đoạn nghiên cứu tiền lâm sàng và lâm sàng. Phương pháp này cho phép dự đoán hoạt tính nhờ sử dụng các mô hình toán học, dự đoán cơ chế tác dụng, cơ chế gây độc của các hợp chất [5].
Phương pháp sàng lọc ảo có thể được chia thành 2 hướng chính bao gồm sàng lọc trên nền tảng hợp chất (LBVS) và sàng lọc trên nền tảng cấu trúc (SBVS). Hướng sàng lọc LBVS sử dụng các dữ liệu tương quan cấu trúc – hoạt tính từ một tệp cơ sở dữ liệu các chất đã biết để lựa chọn chất tiềm năng cho đánh giá thực nghiệm. Hướng nghiên cứu này bao gồm việc tìm kiếm các hợp chất có cấu trúc tương đồng, dẫn xuất, nghiên cứu tương quan cấu trúc – hoạt tính (QSAR) và dược học. Hướng sàng lọc SBVS, theo một cách khác, sử dụng cấu trúc ba chiều của đích sinh học để mô phỏng tương tác ảo với các hợp chất tiềm năng và xếp hạng chúng dựa trên ái lực liên kết hoặc vùng liên kết [6].
Cụ thể trong thời gian gần đây, phần lớn các phác đồ và phương pháp điều trị ung thư đều liên quan đến việc sử dụng hóa trị. Mặc dù có vai trò đáng kể của hóa trị liệu trong việc chữa và kiểm soát ung thư, cơ chế gây độc tế bào của một số tác nhân hóa trị liệu không được đặc trưng rõ ràng. Nhiều loại thuốc hóa trị liệu chống ung thư này sở hữu acid nucleic và các quá trình phụ làm mục tiêu tế bào chính của chúng. Ngoài ra, các nhà nghiên cứu không ngừng nỗ lực để làm sáng tỏ cơ chế chống ung thư cơ bản của thuốc ở cấp độ phân tử bằng cách nghiên cứu phương thức tương tác giữa axit nucleic và thuốc[7, 8]. Ở đây, gắn kết phân tử đóng một vai trò quan trọng trong dự đoán sơ bộ về đặc tính liên kết của thuốc với acid nucleic. Thông tin thu thập được từ kết quả của các cuộc điều tra như vậy rất hữu ích trong việc thiết lập mối tương quan giữa cấu trúc phân tử của thuốc và độc tính tế bào của nó. Hơn nữa, kiến thức này sẽ là công cụ để phát hiện những thay đổi cấu trúc trong một loại thuốc có thể dẫn đến liên kết cụ thể về trình tự/cấu trúc với mục tiêu của chúng (acid nucleic). Sự hiểu biết này có thể được khai thác trong việc thiết kế và tổng hợp hợp lý các loại thuốc mới, sở hữu hiệu quả tốt hơn và giảm tác dụng phụ, do đó liên kết không đặc hiệu hạn chế liều lượng thuốc và tính thường xuyên trong điều trị ung thư [9].
Bài viết tổng quan này đã cung cấp một cái nhìn tổng quan ngắn gọn về tính tiên tiến trong hai phương pháp sàng lọc LBVS và SBVS trong sàng lọc ảo (in silico), nó trở thành một công cụ có giá trị và hiệu quả để tìm kiếm phát triển thuốc mới. Việc sử dụng kết hợp các phương pháp này đã được chứng minh qua các nghiên cứu trên thế giới là đặc biệt hữu ích trong tìm hiểu cơ chế phân tử, tác dụng phụ của thuốc và tái định hướng các dược phẩm để tác dụng mục tiêu qua những đích sinh học mới và điều trị các bệnh khác nhau một cách an toàn.
Lê Thị Thu Trang (viết và tổng hợp)
Nguồn tham khảo:
[1] R. Otávio de Faria, V. R. Moure, M. A. L. d. A. Amazonas, N. Krieger, D. A. J. F. T. Mitchell, and Biotechnology, “The Biotechnological Potential of Mushroom Tyrosinases,” vol. 45, no. 3, 2007. [2] C. M. Song, S. J. Lim, and J. C. J. B. i. b. Tong, “Recent advances in computer-aided drug design,” vol. 10, no. 5, pp. 579-591, 2009. [3] A. J. N. r. D. d. Mullard, “New drugs cost US $2.6 billion to develop,” vol. 13, no. 12, p. 877, 2014. [4] N. S. Pagadala, K. Syed, and J. J. B. r. Tuszynski, “Software for molecular docking: a review,” vol. 9, no. 2, pp. 91-102, 2017. [5] E. Lionta, G. Spyrou, D. K Vassilatis, and Z. J. C. t. i. m. c. Cournia, “Structure-based virtual screening for drug discovery: principles, applications and recent advances,” vol. 14, no. 16, pp. 1923-1938, 2014. [6] P. M. Quân, T. Q. Lê Thị Thùy Hương, P. T. H. M. Toàn, and P. Q. Long, “PHƯƠNG PHÁP SÀNG LỌC ẢO TRONG NGHIÊN CỨU PHÁT TRIỂN THUỐC.” [7] H. J. Lee, W. J. Lee, S. E. Chang, and G.-Y. J. I. j. o. m. s. Lee, “Hesperidin, a popular antioxidant inhibits melanogenesis via Erk1/2 mediated MITF degradation,” vol. 16, no. 8, pp. 18384-18395, 2015. [8] T. Yokozawa, Y. J. J. B. Kim, and P. Bulletin, “Piceatannol inhibits melanogenesis by its antioxidative actions,” vol. 30, no. 11, 2007. [9] L. G. Ferreira, R. N. Dos Santos, G. Oliva, and A. D. Andricopulo, “Molecular docking and structure-based drug design strategies,” Molecules, vol. 20, no. 7, pp. 13384-421, Jul 22 2015.